Dreidimensionale (3D) Konfigurationen von Atomen bestimmen alle Materialeigenschaften. Eine quantitative Vorhersage von 3D-Koordinaten aller Atome und somit deren genauen Gleichgewichtsstrukturen ist eine anspruchsvolle und rechenintensive Aufgabe, die am Anfang praktisch jedes Arbeitsablaufs in der Computerchemie steht. Forscher der Universität Wien haben nun ein neues, auf maschinellem Lernen basierendes Modell entwickelt, um die intensiven Berechnungen abzukürzen und Strukturen direkt aus chemischen Graphen vorherzusagen. Diese neue Methode wird in der aktuellen Ausgabe von Nature Communications vorgestellt.
Chemische Verbindungen werden häufig als starr dargestellt. Tatsächlich sind diese aber flexible dreidimensionale (3D) Objekte, die aus Atomen bestehen, welche sich ständig bewegen und schwingen. Cyrus Levinthal stellte bereits 1969 fest, dass die große Anzahl von Freiheitsgraden chemischer Verbindungen formal zu einer katastrophal großen Anzahl möglicher Konformationen von gut bis zu 10300 führt (Levinthal's Paradoxon). In experimentellen Beobachtungen entsprechen die 3D-Konfigurationen der Atome jedoch wohldefinierten Minima der freien Energie und diktieren damit alle Materialeigenschaften. Das Paradigma, dass die chemische Struktur die Funktion eines Materials bestimmt, ist der Schlüssel für die Entwicklung von Arzneimitteln, für die Optimierung von Katalysatoren oder Reaktionen und Entdeckungen neuer Materialien. Demgemäß wird in den meisten computergestützten Hochdurchsatz-Screening-Kampagnen, einer Methode für schnelle wissenschaftliche Experimente, nur nach den stabilsten Konfigurationen gesucht. Abhängig von der Komplexität der Näherungen, die bei der Abschätzung der Stabilität von Materialien gemacht werden, kann der Aufwand für die Berechnung einer einzigen Struktur von Minuten bis zu Stunden oder sogar Tagen variieren. Angesichts der Größe des Raums der chemischen Verbindungen, der von allen denkbaren Verbindungen gefüllt wird (schätzungsweise mehr als 1060), stellt dieser Kompromiss zwischen Rechenaufwand und Qualität eine große Limitierung im Feld dar.
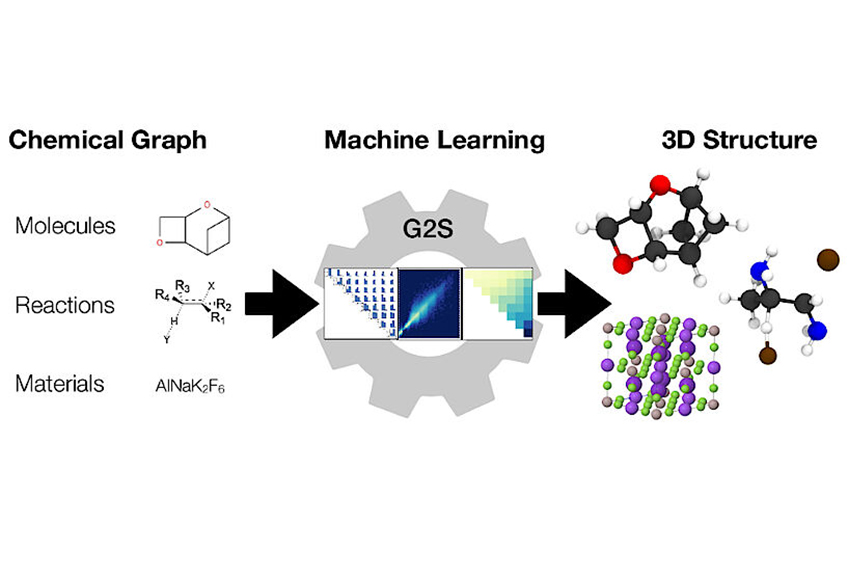
Forscher an der Universität Wien unter der Leitung von Anatole von Lilienfeld haben dieses Problem aus einer anderen Perspektive in Angriff genommen und eine neue Methode entwickelt, die Daten nutzt und universell auf jede Art von Chemie anwendbar ist. Ihre neue Methode, Graph2Structure, verwendet qualitativ hochwertige quantenchemische Daten, um Modelle für maschinelles Lernen zu trainieren. Diese sind in der Lage, neue 3D-Strukturen für chemischen Graphen von ungesehenen Verbindungen vorherzusagen. Diese direkte Zuordnung eines chemischen Graphen zu einer spezifischen 3D-Konfiguration ermöglicht es dem Modell, jede Form der Energieminimierung effektiv zu umgehen, was zu einer Beschleunigung von über einer Million im Vergleich zu den herkömmlichen Methoden führt. "Die Möglichkeit, qualitativ hochwertige Strukturen zu generieren, beschleunigt nicht nur das molekulare Hochdurchsatzdesign, sondern auch den alltäglichen Arbeitsablauf" – so Hauptautor der Studie in Nature Communications Dominik Lemm. "Die zuverlässige Generierung von 3D-Strukturen auch für exotische Chemien, wie Systeme mit offener Schale oder Übergangszustände, ist eine der schwierigsten Aufgaben in der atomistischen Simulation." Weitere Erkenntnisse deuten darauf hin, dass die generierten Strukturen direkt als Input für zusätzliche maschinelle Lernmodelle zur Vorhersage von Materialeigenschaften verwendet werden können. Dadurch wird ein molekularer Graph mit einer strukturabhängigen Eigenschaft auf rigorose und effektivere Weise verknüpft.
Publikation in Nature Communications:
Machine learning based energy-free structure predictions of molecules, transition states, and solids Dominik Lemm, Guido Falk von Rudorff, O. Anatole von Lilienfeld. DOI: 10.1038/s41467-021-24525-7